
 Andreas Meliniotis, Director of Device Development at Vectura, describes how to design inhalation devices to ensure not only satisfactory drug delivery, but also patient compliance with dosing regimens by making such devices easy to use.
Andreas Meliniotis, Director of Device Development at Vectura, describes how to design inhalation devices to ensure not only satisfactory drug delivery, but also patient compliance with dosing regimens by making such devices easy to use.
Some medicines are best delivered via inhalation, often because they are treating a condition that affects the lungs, but with this route of delivery comes the requirement for a suitable device. Traditional inhalers and nebulizers can be difficult to operate and require the patient to time their inspiration breaths with the drug’s release. Additionally, devices may not be appropriate for all patients, such as paediatrics, or may also be difficult to operate for those with dexterity issues, such as osteoarthritis patients.
There is, therefore, a wide scope of opportunity for the development of new, improved devices to ensure inhaled drugs can be accessible for a wide range of patients, which can also treat various diseases and conditions effectively. The starting point when designing a new device is to define its target market, the user needs, and the possible regulatory pathway. Innovators need to work with engineers and designers to focus on what the requirements of both the drug and the patient population are.
First steps
As an initial step, there would usually be a number of different concepts proposed, which are ranked in terms of both benefits and disadvantages. Those that closely match the requirements would be advanced through to the more detailed design stage, where practical considerations such as the manufacturability and scalability are assessed.
The next step is to take these detailed design concepts and make sample devices using 3D printing or another rapid prototyping method. A drawing is no substitute for a physical prototype, which allows a thorough assessment and gives a real sense of how the device might work in the hands of a patient. Having a plastic device also allows tests to be carried out at an early stage as a dry run to reduce time later.
These prototype devices also allow the performance of laboratory tests to determine how the formulated drug is likely to behave in the device, as well as mechanical tests against the key functional parameters defined at the programme’s outset. These assessments will allow the definition of a list of critical quality attributes (CQAs) for the device.
Into plastic
If, after these initial studies, the proposed device looks to be promising, a more robust prototype will be made. Computer-aided design (CAD) and rapid mould tools allow this to be completed typically within six weeks. Having a prototype allows the user needs to be compared against actual functional specifications of a device, which are both testable and can be concept-specific. Table 1 shows examples of how functional specifications relate to user needs.
| User needs | Functional specifications |
| The device shall be operable by the patient population | The device shall require no more than 10N of force to operate the button |
| The device shall be portable | The device shall weigh less than 500g |
| The device shall protect the drug from moisture | The doses shall be contained within individually sealed aluminium blisters |
Table 1. How inhaler device functional specifications relate to user needs.
The next step is device verification, again comparing it to the functional specifications. This will allow a traceability matrix to be created that will collate the evidence that it meets the functional specification and the list of user needs.
The human aspect
Human factors studies are particularly important in the design. Initially, these will be conducted with relatively small numbers of subjects, and with the user groups that are most likely to show user errors, which may include the elderly, where there may be dexterity issues, or young children, who may not have the strength or coordination to operate the device effectively.
It is often easier to recruit healthy volunteers as subjects (or volunteers who are healthy aside from those dexterity issues) in such studies, and these typically highlight potential problems early on in the development process. This allows issues to be eradicated faster and with less expense than to progress a project into late-stage clinical trials only to have it fail because the device was not suitable.
Inhaled drugs exacerbate the challenges with patient compliance because if there is a difficulty in using a device effectively, a patient may think they are taking their medicine, but, in reality, the intended dose of drug is not reaching the lungs properly. The consequence of this, and for all issues around patient compliance, is that potential treatments for patients are stopped or altered because of a perceived lack of efficacy. For device developers, ensuring that a patient is able to use a device both effectively and consistently is vital for improving compliance.
Human factors studies highlight how patients view and interact with a device, and a designer must ensure that the likelihood of poor compliance is reduced as far as possible by good product design, which again must be backed up by success in the human factors studies.
Results from tests are fed back to the device designers, who can refine the design before repeating tests on the new devices. This iterative design-test-feedback-amend process may lengthen the early stages of development, but can ultimately lead to monetary and time savings by establishing an effective device from the outset of drug development.
Understanding how successful the device is in delivering a dose is key, but what human factor studies can also show is whether the devices are being used correctly by patients. What is perhaps most useful is the fact that the studies can indicate exactly how they may be misused, and this could be for reasons such as a minor error or slip by a patient (‘operator error’), a disconnect between a designer’s view on patient intuition when handling a device, or on fundamental misunderstanding of the device concept. Instructions for use (IFU) are provided with the marketed devices, however there is no guarantee that patients will read these, and therefore testing without the IFU can be useful to determine how intuitive the design is and how likely errors are to occur in the market.
Thus, the device development process typically follows a stage gate process, where the project is reviewed with a board of representatives from across the relevant disciplines. The development is structured so that initially the target product profile (TPP) and user requirements, including the user needs and intended uses, are reviewed at the first stage gate, followed by the concepts at the second stage gate, formal designs, device verification, manufacturing equipment, validation and finally preparation for market. The performance of the device continues to be monitored by post-market surveillance to assess whether lifecycle management activities are required. The reviews are conducted with representatives from a number of different disciplines to ensure that every relevant aspect is reviewed prior to continuation through the phases.
Regulatory compliance
Obviously, any device development process must align with the requirements of the regulators, and sufficient evidence of suitability must be included in the registration package. Following the waterfall diagram in the FDA’s 21 CFR Part 820.30 (Figure 1) is a good place to start, and although different regulatory bodies have slightly different requirements, it is important to include an assessment of whether it will meet the demands of the regulators in all the regions where the device is likely to be launched.
Figure 1. FDA-recommended design control parameters for medical device manufacture.
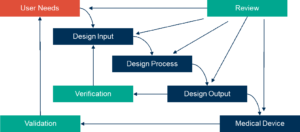
Source: Schematic from 21 CFR 820.30 FDA Design Controls Guidance for Medical Device Manufacturers (March 11, 1997)
Reducing risk
In an ideal scenario, a new product will take advantage of an existing delivery platform, as this affords a degree of proof that the product will be scalable to a commercial process, and issues around scale-up are less likely. Using laboratory-scale manufacturing to explore the design space of the product ahead of large-scale production can help to ensure efficient costs and timescales and also assist in moving the product into the clinic at a relatively early stage without having to invest in the expensive equipment and facilities required for a commercial product in advance.
Applying statistical analysis to test data allows the prediction ofany likely problems once devices are being manufactured and filled at a commercial stage. If any issues are foreseeable, then this analysis will allow measures to be put in place in advance to reduce the potential of large numbers of returns, and the associated cost accompanying poor publicity and loss of reputation.
Various different methods can be applied to reduce the risk. Paper-based exercises such as tolerance analysis can be used to assess the risk, while processes such as failure mode and effects analysis can also help, whether from the manufacturing, design or human factors perspective. Using these outputs to guide development allows risks to be navigated and reduced as a project moves from an idea to a product that is ready for clinical trials and subsequent commercialization.
Validation: a necessary step
Verifying a product against a concept-specific and testable specification is one thing; from this it might be expected that clear values that can be subjected to statistical analysis are generated. Validation, however, requires going all the way back to the original list of user needs and proving that the device meets all those original requirements, not just the specifications that these were used to establish.
Human factors studies provide validation, but patients should be left to use it according to the instructions in the patient instruction leaflet. A clinical trial may also constitute validation that it does what it should.
Ultimately, it is important to remember the person for whom the device is being created: the patient. A device clearly needs to be robust, and deliver the product. But a design that is not patient-centric is unlikely to succeed.
Further Information:
